LA SCLEROSI MULTIPLA OGGI E LA SUA VALUTAZIONE MEDICO-LEGALE
|
Fabrizio Zappaterra primario medico-legale Sede Regionale INPS, TRENTO |
Patrizia Carassai aiuto medico-legale Sede Provinciale INPS, VICENZA |
INTRODUZIONE
Le prime descrizioni di casi di sclerosi multipla (SM) risalgono al 1830 (Cruveilhier, Carswell), ma oltre 160 anni non sono stati sufficienti per chiarire l'eziopatogenesi e quindi definire un protocollo terapeutico decisamente efficace per modificare l'andamento della malattia. Negli ultimi 10 anni si sono compiuti notevoli progressi sulla conoscenza della SM, in base ai quali sono stati approntati trials terapeutici sperimentali al fine di ridurre l'evolutività della malattia.
Benché sia una malattia relativamente a bassa incidenza, la SM è una delle maggiori, se non la principale causa di disabilità neurologica giovanile e della media età e l'INPS è chiamato in causa per il riconoscimento delle prestazioni previdenziali, che hanno una parte considerevole del costo sociale complessivo della malattia stessa.
EPIDEMIOLOGIA
L'epidemiologia osservazionale valuta il "peso" della malattia nella popolazione, la sua distribuzione per età, sesso, località, i fattori di rischio e prognostici.
L'entità di una malattia viene identificata mediante i tassi di mortalità, incidenza e prevalenza per 100.000 abitanti. Così, i tassi di mortalità per SM variano da 0,1 per 100.000/anno in Asia ed Africa, fino a 3 nella Scozia e nell'Irlanda del Nord; l'incidenza da 0,4 per 100.000 nuovi casi/anno nei nativi in Sudafrica, fino a 9,3 nelle isole Orkney della Scozia; la prevalenza infine va dai 4 casi per 100.000 dell'isola di Malta fino ai 309 casi delle isole Orkney (1).
In Italia è stata riscontrata una prevalenza variabile tra 7 e 27 per 100.000 abitanti in larghe popolazioni e tra 30 e 44 in piccole popolazioni. Nella provincia di Trento nel 1978 fu riscontrata una prevalenza di 27 per l'intera popolazione e di 45 nell'U.S.L. Bassa Valsugana (2, 3). A 9 anni di distanza, nel 1987 la prevalenza nell'U.S.L. Vallagarina è risultata di 57 (4), ma non è chiaro se l'aumentata prevalenza sia stata determinata da un aumento del rischio e quindi dell'incidenza, da un affinamento diagnostico, da una più accurata raccolta dei dati o da un prolungamento della vita media degli ammalati.
Già nel 1950, nelle conclusioni di uno studio epidemiologico negli U.S.A., fu avanzata l'ipotesi di un aumento della frequenza con la distanza dall'equatore (5).
Tale ipotesi è stata successivamente confermata anche da una metanalisi di numerosi studi in letteratura (6, 7) che ha indicato un effettivo aumento della prevalenza in base all'aumento della latitudine. Sono state individuate delle fasce geografiche ad alto rischio di malattia (più di 30 casi per 100.000 ab.), come Europa Occidentale sopra il 43° parallelo, Canada, Nord degli U.S.A., Australia del Sud; a medio rischio di malattia (5-25 casi): Europa Orientale, U.S.A. Centrale, Australia Centrale e Settentrionale; a basso rischio di malattia (meno di 5 casi): Asia ed Africa.
Tale mappatura va ad identificare regioni prevalentemente abitate da caucasici e suggerisce l'esistenza di una predisposizione genetica alla SM. Tuttavia, se nordeuropei migrati in Sudafrica e nordamericani migrati alle Haway hanno mantenuto il rischio del Paese originario, i soggetti emigrati prima del quindicesimo anno d'età tendono invece ad assumere il rischio del Paese ospite (1). La malattia quindi sembra in parte condizionata da un fattore esogeno che comincia ad agire prima dei 15 anni, per dare la forma manifesta dopo una latenza di oltre un ventennio.
|
PREVALENZA |
1960 |
1970 |
1980 |
|
Scozia |
- |
106 |
145 |
|
Turku (Finlandia) |
30 |
- |
93 |
|
Hordaland (Norvegia) |
20 |
- |
60 |
|
Ferrara |
10 |
27 |
46 |
|
Tabella 1. Andamento temporale della prevalenza della sclerosi multipla in varie località europee. |
|||
Negli anni, la prevalenza e l'incidenza della malattia (Tabelle 1 e 2) possono essere stabili, come a Rochester (Minnesota) ed a Ferrara (Italia), diminuire come a Rostock (Germania) e nelle isole Orkney (Scozia), aumentare sensibilmente come nella provincia canadese di Newfoundland e Labrador, nella contea norvegese di Hordaland nella regione tedesca della Bassa Sassonia ed in alcune località della Sardegna (1).
|
INCIDENZA |
1950 |
1960 |
1970 |
1980 |
|
Isole Orkney (Scozia) |
10,3 |
10,1 |
7 |
2,2 |
|
Bassa Sassonia |
2,6 |
4,6 |
||
|
Sassari |
2 |
2,6 |
5 |
|
|
Ferrara |
2,1 |
2,1 |
2,4 |
|
|
Rostock (Germania) |
4,5 |
1,8 |
3,7 |
1,8 |
|
Tabella 2. Andamento temporale della incidenza della sclerosi multipla in varie località europee. |
||||
Le caratteristiche climatiche connesse alla latitudine ed all'altitudine, come la temperatura media annuale, le ore di sole giornaliere, le radiazioni cosmiche e le precipitazioni sembrano non influire sull'andamento della SM (8), così come i traumatismi (9) ed il tipo di allattamento (10).
E' stata ipotizzata una relazione fra SM e frequenti viaggi (11), come meccanismo di aumentata esposizione a fattori causali; Amaducci (12) ha segnalato un'associazione fra SM ed esposizione a solventi organici, che altererebbero la barriera ematoencefalica con attivazione del sistema immunitario in soggetti geneticamente predisposti.
IPOTESI EZIOPATOGENETICHE
Le ricerche si sono sviluppate in tre direzioni principali: studi genetici, studi virologici e studi immunologici. Nessuna di esse ha dato dei risultati tali da consentire la definizione di veri fattori eziologici.
Ipotesi genetica
Si è visto come la SM presenti una particolare distribuzione geografica, legata a fattori ambientali condizionati dalla latitudine. A volte però, a parità di latitudine, l'incidenza varia; così è maggiore in Sicilia rispetto a Malta e nei bianchi americani rispetto ai negri. Studi sugli immigrati hanno però rivalutato l'importanza della distribuzione razziale, e quindi dell'influenza in questa malattia di un fattore genetico (1).
E' stato ipotizzato che i geni della suscettibilità alla SM abbiano avuto origine fra i popoli della Scandinavia e della Germania, si siano distribuiti in Europa con le invasioni dei Vichinghi, dei Goti e dei Vandali, e nel Mondo con la colonizzazione da parte degli europei (13, 14); si sarebbero così isolati dei "cluster spaziali" ad altissima incidenza, come nelle isole Orkney del Nord della Scozia.
Molto suggestiva è la distribuzione della malattia fra i vari ceppi razziali del Sudafrica, che, confermata recentemente (15), vede una prevalenza di 57.5 per gli immigrati inglesi, 14.1 per gli inglesi nati in Sudafrica, 10.9 per gli afrikaaners e 3 per la popolazione di colore.
Gli studi sui familiari dei pazienti hanno evidenziato un rischio di oltre 20 volte, rispetto a quello della popolazione generale, per i fratelli; tale rischio cala poi massicciamente fra genitore e figlio e fra cugini (16).
I principali studi sui gemelli finora pubblicati hanno rilevato che risulta concordante il 20-25% delle coppie di gemelli monozigoti ed il 5-9% di quelle dizigoti, indicando chiaramente un ruolo di fattori genetici (1).
In molti gruppi etnici la SM è significativamente associata ad antigeni della regione HLA del cromosoma C6: l'associazione più forte è risultata quella con gli antigeni DR2 e DQw1 delle popolazioni caucasiche (17), ma significativa è anche l'associazione con gli antigeni A3 e B7 nelle popolazioni dell'Europa Occidentale, Centro-Settentrionale e del Canada (18). Dall'altra parte popolazioni con elevata presenza di HLA DR2 possono avere bassa prevalenza di SM, o, viceversa, può essere più stretta l'associazione con il DR4, come per esempio in Sardegna. Ancora, negli studi condotti su casi familiari di SM, non è stata riscontrata una concordanza per il sistema HLA tra i parenti affetti dalla malattia. Tutto questo rende improbabile un coinvolgimento diretto del sistema HLA. E' forse più plausibile un linkage disequilibrium con degli ipotetici geni di suscettibilità, quali quelli del C4 del complemento (19), del Tumor Necrosis Factor o dei geni che codificano per proteine coinvolte nella processazione dell'antigene oppure per il recettore per l'antigene dei linfociti T (T cell receptor: TCR) (20, 21), tutti geni del braccio corto del cromosoma 6.
In alternativa, è anche possibile che geni correlati con il sistema HLA e preposti al controllo della risposta immunitaria, conferiscano alle membrane di particolari popolazioni cellulari una configurazione strutturale tale da agire come entità recettoriale per alcuni virus.
Anche il rapporto 3:2 o 2:1 fra femmine e maschi nella frequenza della malattia (22, 23) è in contrasto con l'ipotesi genetica e propone verosimilmente una interazione fra fattori endocrini e sistema immunitario, in analogia a quanto ipotizzato per altre patologie autoimmuni.
Rimane comunque ancora da definire numero e locazione dei geni che condizionano la suscettibilità segnalata, e ciò sarà possibile, forse, partendo dalla sequenza aminoacidica delle proteine che costituiscono la membrana mielinica.
Ipotesi virale
E' stata formulata già nel 1884 da Pierre Marie; l' isolamento di frammenti virali da tessuti ed il riscontro di anticorpi antivirali nel liquor di pazienti affetti da SM, anche se mai verificato in modo costante, hanno contribuito ad alimentare tale ipotesi.
I pazienti con SM sembrano presentare anticorpi antimorbillo a livelli più elevati rispetto ai controlli (24) ed il morbillo è in grado di causare anche un'encefalite acuta e la panencefalite sclerosante subacuta, che si sviluppa a distanza di diversi anni dall'episodio esantematico. Tuttavia l'aumento di anticorpi antimorbillo si riscontra anche in altre malattie come il LES e l'epatite cronica, per cui risulta un fenomeno aspecifico; inoltre non vi è correlazione fra aumento di anticorpi e bande oligoclonali del liquor ed, infine, la vaccinazione antimorbillosa non sembra abbia influenzato l'andamento della malattia (15).
Alcuni autori hanno poi segnalato una correlazione fra SM ed epidemie di cimurro, che, come è noto, è causato da un paramixovirus come il morbillo. Altri hanno chiamato in causa gli Herpes virus, in particolare quello di Epstein-Barr, anche se la prevalenza di sieropositivi per tali infezioni è talmente elevata nella popolazione generale da rendere difficile una interpretazione del dato come elemento eziopatogenetico (1).
In uno studio, successivamente non suffragato da altre segnalazioni, erano stati riscontrati anticorpi anti virus della scimmia di tipo 5 addirittura nel 56% dei pazienti con SM (25). In passato aveva suscitato particolare interesse anche la segnalazione che 4 dei 7 componenti di un'equipes di ricerca sullo "swayback", una malattia demielinizzante degli agnelli, avevano successivamente sviluppato una SM (26).
Un notevole fermento si è recentemente acceso intorno allo studio dei retrovirus: l'HIV determina nei soggetti con AIDS un'encefalopatia subacuta ed una mielopatia vacuolare, quest'ultima con aspetti simili alla SM (27); l'HTLV-I ha un sicuro neurotropismo e provoca la paraparesi spastica tropicale che si manifesta a distanza di molti anni dall'infezione, ma soltanto in 1 caso su 300 (28), forse per una variabilità della risposta immunologica legata ad una interazione fra virus ed ospite (29).
Vi sono segnalazioni sia pro che contro il riscontro di anticorpi specifici o particelle virali nel siero e nel liquor (15); è recente la segnalazione dell'isolamento di un retrovirus dai leucociti di sangue periferico di pazienti con SM (30).
L'insieme dei dati attualmente disponibili fa propendere per l'ipotesi che non sia implicato un singolo virus nella genesi della malattia, ma che più virus interagiscano dando come risultato ultimo la SM. Potrebbero effettivamente essere in causa gli stessi virus delle comuni malattie infettive, soprattutto se contratte dopo l'adolescenza, virus che dimostrano uno spiccato neurotropismo e sono forse alla base della diversa incidenza della malattia a seconda dell'età di emigrazione da un Paese ad alta ad uno a bassa prevalenza.
La patogenesi virale potrebbe realizzarsi o con la persistenza cronica del virus, consentita da una particolare interazione immunologica con l'ospite, o per l'immunità crociata fra antigeni virali e costituenti della mielina, con l'eventuale comparsa di fenomeni di sensibilizzazione, favoriti questi ultimi da variazioni della barriera emato-encefalica (BEE) post-traumatiche o tossiche.
Recentemente (31) è stata proposta anche l'ipotesi che la SM sia in realtà una malattia tossi-infettiva da difetto di barriera muco-ciliare. Sarebbero implicate le tossine delle Bordetelle (tossine-BB) che costituiscono il più potente attivatore policlonale aspecifico timo-indipendente, di tutte le cellule immunocompetenti, con autotossicità dei linfociti T e dei macrofagi. Le tossine-BB, a causa di un difetto individuale della barriera muco-ciliare (per esempio da deficit congenito di IgA-secretorie o da infiammazioni croniche delle vie respiratorie), passerebbero in modo abnorme nel sangue e determinerebbero le lesioni tipiche della SM con lo stesso meccanismo con cui determinano l'encefalopatia pertussica nel neonato, fisiologicamente privo di IgA-secretorie. Se tale ipotesi fosse verificata, le ripercussioni terapeutiche sarebbero eccezionali, potendo la patologia essere adeguatamente trattata con farmaci di uso comune (antibioticoterapia associata a somministrazione a lungo termine di antistaminici).
Ipotesi immunologica
La sregolazione del sistema immunitario gioca senz'altro un ruolo fondamentale nella eziopatogenesi della SM, benché non si sappia contro quale antigene sia diretta la risposta immunitaria, nè che cosa, eventualmente, abbia portato a tale sregolazione (15).
Nel sangue si riscontra una riduzione dei CD8+ (soppressori) e dei 2H4+, una sottopopolazione dei CD4 che stimola i CD8; sono spesso aumentati i linfociti circolanti e quelli che esprimono markers di attivazione, come il recettore per l'interleuchina-2.
Nel liquor si riscontra una modesta pleiocitosi (5-50 cell./mm3), i linfociti T ed i CD4+ sono aumentati, così come la sintesi intratecale di IgG con 4-5 bande oligoclonali all'elettroforesi. Una piccolissima frazione di questi anticorpi sono diretti contro la proteina basica della mielina (PBM), che però si trova nel liquor anche in altre malattie.
Le esacerbazioni e le fasi di progressione si accompagnano ad un aumento degli anticorpi e della PBM sia in forma libera (soprattutto nelle fasi acute), che in forma legata (soprattutto nelle fasi croniche) (32-34).
Nella genesi della SM potrebbe inserirsi, in soggetti geneticamente predisposti, una anomala attivazione di cellule T e macrofagi (35), normalmente addetti alla sorveglianza del sistema nervoso. Tale anomala attivazione si verificherebbe in risposta ad un fattore esogeno, forse virale, acquisito tra l'infanzia e l'adolescenza (1). La conseguente liberazione di linfochine ed enzimi lisosomiali altererebbe la permeabilità della BEE e la conduzione nervosa, mentre gli anticorpi ed il complemento sarebbero responsabili del danno alle membrane cellulari e alla mielina in particolare (36).
MANIFESTAZIONI CLINICHE
|
SINTOMI D'ESORDIO |
SM STABILIZZATA |
|
Piramidali |
Piramidali |
|
Tronco-encefalici |
Cerebellari |
|
Sensitivi |
Tronco-encefalici |
|
Visivi |
Sensitivi |
|
Cerebellari |
Psico-intellettivi |
|
Urinari |
Facile affaticabilità |
|
Tabella 3. Sintomi più frequenti nella sclerosi multipla. |
|
Come risulta dalla tabella 3, l
a SM può simulare praticamente tutte le sindromi neurologiche (37).a) Disturbi motori: nel 32-41% dei casi l'interessamento piramidale rappresenta l'esordio della malattia ed è presente nel 62% dei pazienti con SM cronico-progressiva. Gli arti inferiori sono coinvolti più frequentemente di quelli superiori, soprattutto all'esordio; la compromissione va da una semplice asimmetria dei riflessi osteotendinei a gravi quadri di paraparesi spastica, è causata soprattutto da demielinizzazione della sostanza bianca del midollo spinale, associata frequentemente a coinvolgimento delle piramidi bulbari, del ponte, dei peduncoli cerebrali e della sostanza bianca dei centri semiovali.
b) Disturbi sensitivi: rappresentano l'esordio della malattia nel 21-55% dei casi e sono presenti nel 52-70% dei pazienti durante l'intero decorso.
All'esordio possono manifestarsi esclusivamente sintomi iperestesici (parestesie, disestesie, dolore), a distribuzione irregolare, successivamente compaiono quadri ipoestesici.
Un tipico sintomo è il segno di Lhermitte: con la flessione del capo si scatena una sensazione di scossa elettrica lungo il dorso e gli arti.
Tali sintomi dipendono più da un coinvolgimento delle fibre mieliniche dei cordoni posteriori del midollo che da interessamento delle vie spino-talamiche.
c) Sintomi tronco-encefalici: il nistagmo, generalmente orizzontale, è presente nel 40-70% dei casi, può essere asintomatico o manifestarsi con episodi di offuscamento della visione o diplopia. L'oftalmoplegia internucleare mono e bilaterale per interessamento del fascicolo longitudinale mediale è estremamente frequente e sintomo quasi patognomonico nei pazienti più giovani. Altre anomalie della motilità oculare estrinseca sono la paralisi dello sguardo orizzontale e verticale da deficit di singoli nervi oculomotori che determinano diplopia.
La disartria sia di tipo pseudobulbare che cerebellare è estremamente frequente soprattutto nei pazienti più disabili e con malattia di più lunga durata.
Disturbi vertiginosi si manifestano soprattutto durante le riacutizzazioni. Rare sono le paralisi complete del VII, VIII e IX nervo cranico.
d) Sintomi visivi: la neurite ottica (NO) è un sintomo d'esordio nel 14-23% dei casi. Si manifesta con un calo del visus, fotofobia e dolore a carico di un occhio; solo nelle fasi più avanzate l'interessamento è bilaterale, ma la cecità è rara e talvolta il rilievo è solo strumentale, senza sintomi visivi.
Obiettivamente si può evidenziare un calo del visus con eventuali scotomi; il FOO può risultare nella norma (NO retrobulbare) o evidenziare edema della papilla con emorragie ed essudati che possono evolvere fino all'atrofia ottica.
e) Sintomi cerebellari: l'atassia dinamica rappresenta l'esordio della SM nel 13% dei casi, colpisce il 50% dei pazienti in fase cronica e può essere il principale fattore di disabilità.
f) Disturbi psico-intellettivi: interessano il 54-65% dei pazienti e sono inquadrabili come disfunzioni cognitive di tipo sottocorticale con alterazioni delle capacità mnesiche e del ragionamento concettuale ed astratto; sono correlati con l'entità delle anomalie encefaliche evidenziate alla risonanza magnetica, nelle forme di più lunga durata e di maggiore disabilità.
Dal punto di vista psichiatrico, turbe debressivo-euforiche sono riscontrabili nel 75% dei casi.
g) Affaticabilità: è uno dei sintomi maggiormente invalidanti, presente nel 78% dei casi.
h) Sintomi genito-urinari e sfinterici: sono riscontrabili nel 78% dei pazienti, rappresentati da pollachiuria, minzione imperiosa ed incontinenza, stipsi, talvolta urgenza a defecare o vera incontinenza fecale. Possibile è la dissinergia vescico-uretrale che determina ristagno vescicale, reflusso vescico-ureterale ed infezioni ricorrenti delle vie urinarie superiori.
Associata ai disturbi della minzione è relativamente frequente l'impotenza sessuale nel maschio.
i) Sintomi parossistici: il più frequente è la nevralgia del trigemino, spesso bilaterale; seguono la disartria parossistica e le crisi epilettiche generalmente parziali (tipicamente a carico dell'arto superiore).
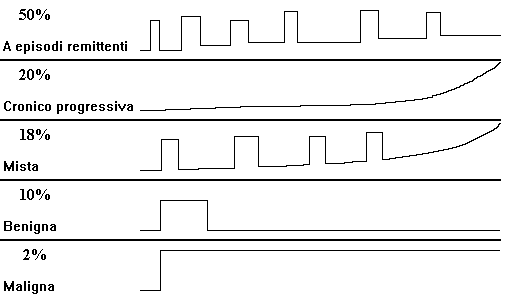
DECORSO
All'inizio (figura 1) la SM è caratterizzata da un andamento a ricadute e remissioni più o meno complete. Il primo attacco solitamente si risolve completamente o quasi, mentre le ricadute durano alcune ore o alcuni giorni ed il recupero richiede settimane o mesi; il tasso di ricadute è stato valutato tra 0.1 e 0.85 attacchi/anno.
|
Figura 1. Schematizzazione del decorso della SM. |
La fase cronico-progressiva caratterizza l'esordio della malattia nel 9-37% dei casi, oppure rappresenta l'evoluzione della forma remittente. Consiste in un lento e progressivo aggravamento della condizione clinica con riacutizzazioni (forma mista) o senza; comporta un accumulo di disabilità da 0,3 a 1,5 punti alla scala di Kurtzke in 5 anni, a seconda della disabilità iniziale (vedi allegato 1).
L'andamento benigno è caratterizzato da 1 o 2 episodi acuti, seguiti da una remissione praticamente completa che dura indefinitivamente; nella forma maligna, viceversa, già il primo episodio determina dei danni neurologici gravissimi e praticamente irreversibili.
Complessivamente l'11-34% dei pazienti non è più in grado di lavorare dopo 15 anni dall'esordio (38, 39), il tasso di sopravvivenza a 25 anni è pari al 75%, rispetto ad un tasso atteso pari all'86%.
Sembra che l'andamento nei primi 5 anni sia indicativo anche dell'andamento successivo (40). La prognosi è migliore nel sesso femminile, ad insorgenza in età giovanile, con inizio acuto seguito da remissione e con sintomi iniziali di tipo sensitivo; è peggiore nel sesso maschile, ad insorgenza dopo i 35-40 anni, a decorso cronico-progressivo, con sintomi iniziali piramidali o cerebellari.
E' un fattore di aggravamento l'innalzamento della temperatura corporea da stati febbrili, esercizio fisico, bagni caldi, esposizione solare: è tipico il "fenomeno di Uhthoff", cioè l'annebbiamento visivo da blocco di conduzione in assoni parzialmente demielinizzati. La gravidanza sembra avere un ruolo protettivo, ma il successivo puerperio è un sicuro fattore di aggravamento. Le infezioni virali facilitano la comparsa di ricadute, mentre ciò non è dimostrato per i traumi, gli stresses psico-fisici, gli interventi chirurgici e l'anestesia.
DIAGNOSI
Dopo lo schema proposto da Charcot nella prima descrizione della malattia (41), la classificazione diagnostica più utilizzata a livello internazionale è stata quella proposta da Shumacher e al. (42) che considerando i dati anamnestici e l'obiettività neurologica, classificava la SM come definita, probabile e possibile a seconda di quanti dei seguenti criteri venivano soddisfatti:
1. età di esordio tra i 10 ed i 50 anni;
2. presenza di segni neurologici obiettivabili;
3. sintomi e segni neurologici compatibili con sofferenza della sostanza bianca del S.N.C.;
4. disseminazione temporale:
a) due o più attacchi della malattia (> 24 ore) separati da un intervallo di almeno un mese;
b) progressione di sintomi e segni per almeno 6 mesi;
5. disseminazione spaziale: coinvolgimento di due o più aree contigue;
6. nessuna spiegazione alternativa.
Su analoghi elementi è basata la classificazione di McDonald e Halliday del 1977 (43) che suddivide la SM in tre livelli di certezza diagnostica: definita, probabile e sospetta.
La diagnosi differenziale va posta con le malattie che provocano lesioni multiple del S.N.C., quali LES, m. di Sjöegren, m. di Lyme, eredoatassia, leucodistrofia, tumori (gliomi) del tronco encefalico, linfomi primitivi del S.N.C., malformazioni artero-venose, cisti aracnoidee.
Dal punto di vista diagnostico, il primo salto di qualità è stato fornito dall'introduzione in campo diagnostico della tomografia assiale computerizzata (TAC), in grado di evidenziare aree di ipodensità soprattutto nelle regioni periventricolari, corrispondenti istologicamente a placche di demielinizzazione, oppure un franco quadro di atrofia cerebrale nelle forme di più lunga durata.
La TAC con mezzo di contrasto ha permesso poi di dimostrare aree di accumulo, espressione di danno della BEE, presenti soprattutto durante le fasi di attività della malattia e che tendono a scomparire dopo trattamento steroideo.
L'introduzione della tecnica dei potenziali evocati prima e della risonanza magnetica nucleare poi, ha ulteriormente affinato la capacità diagnostica strumentale.
Risonanza magnetica nucleare (RMN).
Introdotto nel 1973, si è dimostrato l'esame strumentale più utile per la diagnosi di SM, sostituendo completamente la TAC (44). Le alterazioni rilevabili alla RMN sono espressione di un modificato contenuto idrico, quindi aspecifiche, ma rilevabili nel 95-98% delle forme clinicamente definite di SM, con minor frequenza nelle forme probabili e sospette della malattia (80% e 50% rispettivamente); corrispondono alle placche di demielinizzazione, tipiche della malattia.
Le aree di alterato segnale sono preferenzialmente distribuite intorno ai ventricoli, nel corpo calloso e nel ponte, sono prevalentemente multiple, talvolta confluenti.
La RMN consente di dimostrare una disseminazione spaziale delle lesioni, anche nei casi non sospettabili clinicamente (45), oppure nelle forme sospette per SM, come la NO isolata (46) o la mielopatia ad eziologia ignota (47). La diagnosi di SM definita va posta comunque solo dopo uno studio clinico/strumentale longitudinale che dimostri anche la disseminazione nel tempo delle lesioni (46,48).
Nella maggior parte dei pazienti, spesso in fase clinica silente, compaiono delle nuove aree di lesione, mentre altre rimangono invariate, si ampliano, si riducono o addirittura scompaiono, sia nella forma remittente, che in quella cronica progressiva.
La comparsa di nuove lesioni è dieci volte più frequente delle ricadute cliniche.
Le nuove lesioni aumentano di volume in 2-4 settimane e si riducono per 3-6 settimane. Non è raro osservare comportamenti opposti contemporanei nello stesso paziente, a suggerire che le riacutizzazioni non sono necessariamente scatenate da cause esterne o interne, ma che anche fattori locali influenzano l'evoluzione delle lesioni (37).
La RMN dopo somministrazione endovena di un mezzo di contrasto paramagnetico, l'Acido gadolinio-dietilentriamino pentacetico (Gd-DTPA), permette di evidenziare lesioni indicative di una alterazione locale della BEE, che possono precedere di 1-2 settimane la comparsa di aree di alterato segnale alla RMN convenzionale ed i segni clinici di riacutizzazione (49-51).
La captazione di Gd-DTPA si verifica sia in nuove che in vecchie lesioni in fase di riaccensione, dura solo 3-8 settimane, e rappresenta un indubbio segno di attività di malattia.
L'uso del Gd-DTPA non migliora la scarsa sensibilità della RMN per lesioni del midollo spinale (52), ma permette di evidenziare lesioni a livello del nervo ottico; con questa tecnica è stato possibile dimostrare che le aree di accumulo sono notevolmente inferiori di frequenza nelle SM cronico-progressive primarie, rispetto a quelle secondarie ad una prima fase remittente, quasi che le due forme avessero elementi patogenetici differenti (53,54).
Periodi di follow-up con valutazioni mensili o bimensili delle placche con RMN e Gd-DTPA danno risultati estremamente più attendibili sull'efficacia della terapia steroidea o immunosoppressiva, rispetto ai trials clinici che si pongono come end-point la frequenza delle poussées cliniche (37).
Potenziali evocati (PE)
La base fisiopatologica delle alterazioni riscontrate con i PE nella SM è la demielinizzazione, che determina un rallentamento della conduzione degli impulsi lungo le vie sensoriali testate, comporta un aumento di latenza delle risposte evocate o, nei casi più gravi, una disgregazione morfologica o addirittura l'assenza del potenziale stesso.
Introdotta nei laboratori di neurofisiologia verso la fine degli anni '60, questa metodica rimane ancora uno strumento insostituibile per la valutazione dei pazienti con la SM in quanto:
1) fornisce informazioni sulla funzionalità di specifiche vie nervose non ottenibili con altri mezzi (la RMN tradizionale non è in grado di differenziare l'edema dalla demielinizzazione e dalla gliosi, che hanno effetti diversi sulla conduzione nervosa);
2) studiano anche aree del S.N.C. ancora mal visualizzabili con la RMN, quali il nervo ottico ed il midollo spinale;
3) talvolta risultano alterati anche in pazienti con RMN negativa o con lesioni singole.
Nelle forme di SM definita, l'uso combinato di tutti i PE (visivi-PEV, somatosensoriali-PESS, acustici del tronco encefalico-PEA), permette di rilevare alterazioni con frequenze di poco inferiori alla RMN; nel 30-50% dei pazienti sono presenti anche in fase subclinica (55).
La recente introduzione delle metodiche di stimolazione corticale permette di indagare neurofisiologicamente anche le vie motorie centrali. In uno studio (56) i PE motori (PEM) sono risultati alterati nel 94% delle forme di SM definita e nel 44% delle sospette. I PEV, tuttavia, appaiono più sensibili dei PEM nel riconoscere alterazioni subcliniche delle vie di conduzione.
I PE sono risultati utili per aumentare il livello di certezza diagnostica delle forme sospette o iniziali o per predire il successivo decorso clinico (alto rischio di aggravamento con PE alterati), anche se con risultati inferiori rispetto alla RMN (37).
Una volta alterati i PE tendono a rimanere tali; la mancata comparsa di modificazioni a distanza di tempo viene in genere ascritta all'efficacia della terapia, per cui sono stati utilizzati nel follow-up di trattamenti con azatioprina, globulina antilinfocitaria e steroidi con risultati positivi. Il loro utilizzo nel monitoraggio della terapia steroidea ad alte dosi nelle fasi di acuzie ha invece conseguito solo risultati negativi.
Liquido cefalorachidiano (LCR)
La dimostrazione di particolari alterazioni del LCR consente di porre diagnosi di SM definita "laboratory supported" (56), analoga, dal punto di vista diagnostico, alla SM clinicamente definita.
Le incostanti alterazioni delle cellule liquorali sono risultate di scarsa utilità clinica; viceversa particolarmente utili si sono rivelate le cosiddette bande oligoclonali, l'IgG index e la proteina basica della mielina (PBM).
- Bande oligoclonali: l'incremento assoluto e/o relativo (rispetto all'albumina) delle IgG liquorali, si associa a modificazioni qualitative che danno luogo alla separazione elettroforetica in bande oligoclonali; sembra derivino dall'attivazione anomala di determinati cloni linfocitari, probabilmente implicati nell'autoaggressione della mielina, anche se al momento attuale non è stato possibile evidenziare una loro specifica attività anticorpale.
- IgG index: è determinato dal confronto fra il rapporto IgG / albumuna nel LCR e nel siero; se risulta superiore a 0,7 è espressione di un'aumentata sintesi liquorale di IgG.
Tali reperti non hanno un chiaro significato prognostico nelle forme definite di malattia, ma aumentano la certezza diagnostica delle forme sospette e costituiscono un indice di rischio di sviluppare ulteriori episodi neurologici. Analogo significato riveste il riscontro di BO costituite da IgM (57), mentre l'aumentata sintesi intratecale di IgD sembra correlarsi all'attività di malattia nella SM a episodi remittenti (58).
Altre alterazioni qualitative sono ancora in fase di studio come l'incremento del rapporto delle catene leggere kappa/lambda e la dimostrazione di catene leggere kappa libere.
- PBM: viene rilasciata nel LCR a seguito di demielinizzazione acuta, costituisce un marker di attività di malattia e sembra correlarsi con il successivo decorso della SM e con gli effetti terapeutici da trattamento steroideo od immunosoppressivo.
Eziopatogenesi delle lesioni
Qualunque sia la causa scatenante, l'alterazione più precoce è rappresentata da un aumento della permeabilità della BEE, che può precedere l'insorgenza dei sintomi, ma non l'alterazione dei PE probabilmente legata alla demielinizzazione. Segue l'edema, quindi il ripristino in circa un mese della normale permeabilità della BEE e la risoluzione dell'edema che lascia una piccola cicatrice residua. Il processo può ripetersi ai margini delle lesioni più grandi che si espandono in modo centrifugo (59).
I sintomi ed i segni della fase acuta si verificano durante la fase dell'infiammazione, che pare dunque svolgere un ruolo importante nella fisiopatologia della malattia attraverso la mediazione delle citochine, che hanno un notevole effetto sull'eccitabilità di membrana. La riduzione delle citochine al termine della fase infiammatoria permetterebbe il ripristino della conduzione pur permanendo la demielinizzazione, a meno che un'importante reazione gliale o la perdita assonale non abbiano condotto a deficit neurologici
TERAPIA
L'ipotesi eziopatogenetica che considera la SM scatenata da un processo infettivo contratto in giovane età ed in soggetti geneticamente predisposti, su cui si innesta una reazione immunitaria rivolta forse contro la mielina è alla base della ratio di terapie immunosoppressive, desensibilizzanti, immunomodulatrici ed antivirali.
Utilizzate singolarmente o in associazione, è ben noto come nessuna abbia a tutt'oggi dato risultati soddisfacenti a distanza (60). Inoltre nei trials terapeutici meno recenti la variabilità clinica della SM e la mancanza di un metodo univoco per valutare lo stato di attività della malattia sono stati i principali fattori di confondimento per una corretta interpretazione dei risultati.
L'applicazione delle attuali metodiche strumentali e di laboratorio hanno permesso e permetteranno in futuro il reclutamento di malati omogenei ed in fasi più precoci di malattia per una maggiore certezza dei risultati terapeutici.
Allo stato attuale l'ACTH e i corticosteroidi rappresentano l'unica terapia universalmente accettata, per brevi periodi di tempo, al fine di ridurre la durata e la gravità delle singole ricadute, senza peraltro modificare né il grado di recupero né l'andamento della malattia (37).
Risultati contrastanti, parziali o del tutto deludenti, ancorché gravati da tutt'altro che irrilevanti effetti collaterali, hanno dato la Ciclofosfamide, l'Azatioprina, la Ciclosporina A, l'irradiazione linfoide totale, la terapia desensibilizzante, gli immunomodulatori, la plasmaferesi, l'ossigenazione iperbarica, gli acidi linoleico e linolenico.
L'attenzione attuale è rivolta a trials a lungo termine con l'uso di interferoni, noti antivirali che possono ridurre le esacerbazioni della SM (60, 61).
Speranze di nuovi orizzonti terapeutici vengono da tecniche immunologiche sofisticate, come l'uso di anticorpi anti molecole di adesione, che impediscono alle cellule leucocitarie di superare la BEE e di danneggiare la mielina e si sono dimostrati efficaci nella prevenzione della encefalomielite allergica (62).
RIABILITAZIONE NELLA SM
In assenza di una terapia causale, la riabilitazione ha assunto nell'ambito della SM un ruolo fondamentale nel consentire al paziente il grado massimo di autonomia possibile, opponendosi allo sviluppo e alla progressione di severe complicanze, fortemente inabilitanti, come per esempio la spasticità (63, 64).
La terapia riabilitativa deve essere multidisciplinare, non limitandosi cioè ai soli aspetti fisici, ma anche psichici, sociali, relazionali ed occupazionali. Deve coinvolgere non solo i livelli neurologici spinali e troncoencefalici, ma anche le più alte funzioni cerebrali.
Per quanto attiene la fisioterapia, la stessa va personalizzata sia all'individuo che alla forma e fase clinica della malattia. A titolo esemplificativo, durante gli episodi acuti sono utili, per mantenere la mobilizzazione e ridurre la spasticità, gli esercizi fisici passivi che vanno poi sostituiti con la partecipazione attiva del paziente nelle fasi di recupero. La mobilizzazione deve essere comunque sempre personalizzata, sotto la supervisione del fisiatra e del fisioterapista, evitando la pratica di esercizi fisici pesanti che possono invece peggiorare il senso di debolezza muscolare (60).
Nonostante alcuni autori sostengano tuttora che nelle fasi iniziali sia sufficiente un supporto rieducativo solo dopo le esacerbazioni, nella situazione ideale la riabilitazione andrebbe inserita come una normale abitudine quotidiana del paziente, sin dall'esordio della malattia stessa (64).
Per incrementare il grado di autonomia del paziente può essere utile anche addestrarlo all'uso di ausili ortopedici o alla pratica dell'autocateterismo vescicale.
CONSIDERAZIONI MEDICO-LEGALI CONCLUSIVE
Benché il medico legale si trovi ad operare in differenti situazioni valutative, alla base di ciascuna di esse esiste un fattore comune rappresentato dal danno biologico, inteso esclusivamente come alterazione dell'integrità psicofisica del soggetto (danno evento). Esso prescinde dalle ripercussioni che il danno stesso ha sull'ambiente relazionale, sociale, occupazionale, produttivo del singolo individuo e che meglio sono inquadrabili nel "danno conseguenza" rappresentato dal danno alla salute. E' evidente come il "danno evento" costituisce l'unica effettiva base comune ad ogni tipo di valutazione medico-legale e che, come tale, può trovare una concordanza valutativa universale esprimibile anche in percentuali rigorosamente tabellate. Non altrettanto si può dire del "danno conseguenza" che, invece, proprio per sua definizione, richiede una valutazione personalizzata e quindi non tabellabile.
Partendo da questi presupposti e riferendoli alla SM, esistono delle peculiarità insite nella malattia che debbono essere tenute presenti dal medico legale, in qualsiasi ambito valutativo si trovi ad operare:
1) Incertezza diagnostica: singoli episodi di NO, encefalite o mielite, soprattutto se a lesione unica, non sono necessariamente espressione di SM; una valutazione effettuata in fase molto precoce di malattia, annulla un importante criterio diagnostico della SM che è rappresentato dalla disseminazione temporale; le stesse definizioni di "leucoencefalomielite" o "leucodistrofia", talvolta usate per non rivelare l'esatta natura della patologia al paziente, in altre occasioni sottintendono un'incertezza diagnostica o addirittura una diagnosi completamente diversa dalla SM.
2) Certezza diagnostica con esiti neurologici in evoluzione: in caso di SM definita o probabile, il quadro neurologico che risulta dalla cartella clinica, può essere anche completamente reversibile nell'arco di pochi mesi e pertanto: a) è indispensabile la visita dell'interessato, evitando la definizione agli atti; b) i deficit neurologici rilevati non hanno ancora il requisito della permanenza, per cui ogni conclusione andrebbe rimandata di alcuni mesi.
3) Certezza diagnostica con esiti neurologici stabilizzati: è la situazione in cui la valutazione del danno biologico risulta più agevole, almeno negli ambiti valutativi previdenziali ed assistenziali in cui, anche ogni eventuale successivo aggravamento può trovare il suo giusto riconoscimento valutativo.
Avendo ben chiari questi elementi, è dunque possibile affrontare il problema della valutazione percentuale del danno biologico da SM. La scala funzionale e di invalidità di Kurtzke e la scala di invalidità espansa EDSS (allegati 1, 2, 3), pur nate con finalità essenzialmente di tipo clinico e riabilitativo e quindi con i limiti di dover poggiare, anche se molto marginalmente, su dati soggettivi riferiti e che risentono comunque del vissuto psichico del paziente, risultano di particolare utilità anche per un uniforme inquadramento medico legale delle disabilità legate alla malattia.
Su questa base, è possibile individuare cinque gruppi di pazienti. Le valutazioni sono riferite, ovviamente, ai casi di diagnosi certa, perciò, anche a fronte di deficit neurologici completamente regrediti, la valutazione medico legale non può ignorare il problema dei fattori prognostici legati all'evoluzione della malattia stessa: da qui l'assenza, in questa proposta, della valutazione 0 in merito al danno biologico da SM. Nell'ambito dei primi due gruppi individuati, il "peso" di questi fattori prognostici non deve, comunque, meramente tradursi in una maggiorazione valutativa aprioristica che ricorderebbe assai dappresso l'antica quota di solidarietà "etica"; può, invece, far oscillare di qualche punto la valutazione, sempre all'interno del range proposto, qualora siano ravvisabili alcuni elementi individuati sulla base di ampi studi epidemiologici. E' pur vero che questi non consentono di predire con certezza l'andamento della malattia nel singolo paziente, ma va ricordato che (37) l'evoluzione della SM nei primi 5 anni è ritenuta estremamente indicativa dell'andamento successivo, così come il tipo di sistema funzionale interessato all'esordio (prognosi favorevole per un esordio con sintomi sensitivi, sfavorevole se piramidali o cerebellari) e l'età d'insorgenza (evoluzione più rapida dopo i 40 anni).
|
GRADO EDSS |
DANNO BIOLOGICO |
MOTIVAZIONI |
|
0.0 / 2.5 |
10 £ 20 % |
Soggetti senza deficit neuromotori o con minima disabilità coinvolgente uno o più sistemi funzionali (FS). |
|
3.0 / 4.0 |
> 20 £ 40 % |
Soggetti autonomi ed autosufficienti, ma con disabilità che coinvolgono uno o più FS in misura variabile da moderata a relativamente marcata. |
|
4.5 / 5.0 |
> 40 £ 60 % |
Soggetti che presentano compromissioni funzionali da relativamente gravi a gravi, ancora capaci di deambulare autonomamente, ma per tratti limitati. |
|
5.5 / 6.0 |
> 60 £ 80 % |
Soggetti che necessitano di supporti per deambulare o che hanno un' autonomia locomotoria molto limitata. |
|
6.5 / 9.5 |
> 80 % |
Soggetti non autosufficienti, con vari gradi di grave limitazione sino all'allettamento e alla totale dipendenza da altri. Come ormai pressoché unanimemente accettato, il danno biologico del 100% corrisponde alla morte del paziente (grado 10.0 della scala EDSS). |
Rimanendo sempre in tema di valutazione del danno, ma passando all' ambito valutativo INPS, è ben vero che il danno qui tutelato non è quello biologico, ma questo costituisce il presupposto omogeneizzabile e la base su cui si fonda la successiva definizione valutativa del "danno conseguenza", nel caso di specie rappresentato dalla riduzione della capacità lavorativa in occupazioni confacenti. Tenendo quindi presenti i presupposti del danno biologico da SM, si può orientativamente (e non certo tassativamente vista la necessità di personalizzare il danno conseguenza) individuare la fascia di invalidità pensionabile (Art. 1 L.222/84) dal grado 4 al grado 5 della scala EDSS che corrispondono ad un danno biologico da noi valutato tra il 40 ed il 60%. Occorre comunque sempre valutare attentamente la compatibilità degli esiti neurologici con l'attività lavorativa confacente, stante anche l'importanza della terapia occupazionale, sia per superare l'inevitabile ripercussione psichica, sia per mantenere quanto più possibile l'inserimento sociale del malato. A nessuno può sfuggire come l'integrazione delle competenze medico legali previdenziali ed assistenziali consentirebbe ( per esempio nel caso di non raggiungimento dei requisiti previsti dall'art.1) l'immediata pronuncia in merito ad una percentuale di invalidità utile, ad esempio, per un collocamento lavorativo protetto, senza costringere l'interessato a presentare nuove domande, ad affrontare nuovi e talora lunghi iter amministrativi, a sottoporsi a nuovi accertamenti medici.
Quando la disabilità raggiunge o supera il grado 5.5 della scala EDSS (danno biologico >60%), l'assicurato è da considerarsi inabile (Art. 2 L.222/84).
Con una disabilità pari o superiore al grado 6.5 EDSS, il paziente necessita di assistenza continuativa (danno biologico >80%).
RIASSUNTO
La sclerosi multipla è una malattia, a probabile genesi autoimmunitaria, della sostanza bianca del S.N.C., che comporta fenomeni di edema, infiammazione e gliosi a carico dell'encefalo, del midollo spinale e del nervo ottico, responsabili dell'estrema variabilità delle manifestazioni d'esordio, delle riacutizzazioni e del decorso della malattia.
Da tale variabilità è dipeso, soprattutto nel passato, l'estrema incertezza diagnostica alle prime fasi della malattia, superata in parte attualmente da sofisticate metodiche d'indagine.
Anche l'efficacia delle terapie sperimentate è di difficile interpretazione per il decorso irregolare degli episodi nei singoli pazienti.
E' una malattia praticamente ubiquitaria nel mondo, sebbene a diversa prevalenza; ha una preferenza per il sesso femminile e colpisce soggetti di età medio-giovane, portando nella maggioranza dei casi ad una disabilità rilevante ed interessando quindi l'Istituto Nazionale di Previdenza Sociale per il riconoscimento delle dovute prestazioni.
BIBLIOGRAFIA
1) Rossi G.:"Epidemiologia della sclerosi multipla". Atti del Convegno "Approccio pratico alla sclerosi multipla". Ex Arte Salus -Società Medico-Chirurgica Roveretana, 1992, 178:3-10.
2) Rossi G., Ferrari G.:"Sclerosi multipla in provincia di Trento nel 1977: studio epidemiologico" in Ferrari G.:"Epidemiologia per la prevenzione delle malattie neurologiche". Verona, Ed. Libreria Cortina, 1980.
3) Rossi G., Ferrari G., Dal Ri E.:"Sclerosi multipla in provincia di Trento nel 1979: ricerca epidemiologica". in Atti del 2° Convegno Nazionale di Neuroepidemiologia, Milano 12-13 dic. 1980.
4) Moresco M., Rossi G.:"Studio di prevalenza della sclerosi multipla nel 1987 nella U.S.L. Vallagarina - Rovereto". Atti del Convegno "Approccio pratico alla sclerosi multipla". Ex Arte Salus -Società Medico-Chirurgica Roveretana, 1992, 178:10-12.
5) Limburg C.C.:"The geographic distribution of multiple sclerosis and its estimated prevalence in the United States". Proceedings of the Association for Research into Nervous and Mental Diseases, 1950, 28:15-24.
6) Kurtzke J.F.:"Epidemiology of multiple sclerosis" in Hallpike J.F., Adams C.W.M., Tourtelotte W.W. (eds):"Multiple Sclerosis", London, Chapman and Hall, 1983.
7) Kurtzke J.F.:"Epidemiology of Multiple Sclerosis" in Vinken, Bruyn, Klawans, Koester (ed.):"Handbook of clinical Neurology", 1985, 47:259-87.
8) Norman J.E., Kurtzke J.F., Beebe G.W.:"Epidemiology of Multiple Sclerosis in US veterans. 2. Latitude, climate and risk of Multiple Sclerosis". J. Chronic Dis., 1983, 36:551-559.
9) Bamford C.R., Sibley W.A., ThiesC.:"Trauma as an etiologic and agravating factor in multiple sclerosis". Neurology, 1981, 31:1229-1234.
10) Spencely M., Dick G.:"Breast-feeding and multiple sclerosis". Neuroepidemiology, 1982, 1:216-222.
11) Visscher B.R., Bunnel D.H., Detels R.:"Multiple sclerosis and multiple moves: an etiologic hypothesis". Am. J. of Epidemiology, 1981, 113:140-143.
12) Amaducci L., Arfaioli C., Inzitari D., Marchi M.:"Correlazione tra agenti tossici esogeni e S.M.: studio epidemiologico e clinico". Atti del II Convegno Nazionale di Neuroepidemiologia, Milano, 1980, 178-181.
13) Poser C.M.:"Multiple sclerosis in tropical and subtropical countries" in Roman G., Toro G. (eds.):"Tropical Neurology". Florida, CRC press, 1990.
14) Poser C.M. :"The dissemination of multiple sclerosis: a Viking saga? A hystorical essay". Ann.Neurol.,1994, 36(S2):S231-S243.
15) Comi G.:"Ipotesi eziopatogenetiche per la sclerosi multipla". Atti del Convegno "Approccio pratico alla sclerosi multipla". Ex Arte Salus - Società Medico-Chirurgica Roveretana, 1992, 178:12-18.
16) Sadovnick A.D., McLeod M.J.:"The familial nature of multiple sclerosis - Empiric recurrence risk for first - second - and third degree relatives of patients". Neurology, 1981, 31:1039-1041.
17) McFarland H.F.:"Genetic influences in multiple sclerosis". An Update on Multiple Sclerosis (a cura di Battaglia M.A.), 1988, 4:7.
18) Myrianthopulos N.C.:"Genetic aspects of multiple sclerosis" in Vinken, Bruyn, Klawans, Koetsier (eds.):"Handbook of Clinical Neurology", 1985, 47, 3:289-317.
19) Marcelli-Barge A., Poirier J.C.,Chantome R. e coll.:"C4AQ0 and HLA-DR2, risk factors in multiple sclerosis". Res. Immunol., 1990, 141:739-741.
20) Seboun E., Robinson M.A., Doolittle D.H. e coll.:"A susceptibility locus for multiple sclerosis is linked to the T cell receptor beta chain complex". Cell, 1989, 57:1095-1100.
21) Fieschi C., Salvetti M., Buttinelli C. e coll.:"Sclerosi multipla: virus e geni della risposta immunitaria". Fed. Med., 1992, 3:15-17.
22) Acheson E.D.:"Epidemiology of multiple sclerosis". Brit. Med. Bull., 1977, 33:9-14.
23) Kurtzke J.F., Beebe G.W., Norman J.E.:"Epidemiology of multiple sclerosis in US veterans. 1. Race, sex and geographic distribution". Neurology, 1979, 29:1228-1235.
24) Compston D.A.S., Vakarelis B.N., Paul E.:"Viral infection in patients with multiple sclerosis and HLA-DR matched controls". Brain,1986, 109:325-344.
25) Goswami K.K.A., Randall R.E., Lange L.S., Russel W.C.:"Antibodies against the paramyxovirus SV5 in the cerebrospinal fluids of some multiple sclerosis patients". Nature, 1987, 327:244-247.
26) Campbell A.M.G., Daniel P., Porter R.J.:"Disease of the nervous system occurring among research workers on swayback in Lambs". Brain, 1947, 70:50-58.
27) Shaw G.M., Harper M.E., Hahn B.H. e coll.:"HTLV-III infection in brains of children and adults with AIDS encephalopathy". Science, 1985, 227:177-182.
28) Gessain A., Barin F., Vernant J.C. e coll.:"Antibodies to human T-lymphotropic virus type-1 in patients with tropical spastic paraparesis". Lancet, 1985, 2:407-410.
29) Usuku K., Sonoda S., Osame M. e coll.:"HLA haplotype-linked high immune responsiveness against HTLV-I in HTLV-I associated myelopathy: comparison with adult T-cell leukemia/lymphoma". Ann. Neurol., 1988, 23(suppl.):S143-S150.
30) Perron H., Lalande B., Gratacap B e coll.:"Isolation of a retrovirus from patients with multiple sclerosis". Lancet, 1991, 337:862-863.
31) Fiore D.:"Una malattia tossi-infettiva da difetto di barriera: la sclerosi multipla". Abstract in "Il medico d'Italia, 1995, 25:13.
32) Catz I, Warren K.G.:"Intrathecal synthesis of autoantibodies to myelin basic protein in multiple sclerosis". Can. J. Neurol. Sci., 1986, 13:21-24.
33) Warren K.G., Catz I.:"A myelin basic protein antibody cascade in purified IgG from cerebrospinal fluid of multiple sclerosis patients". J. Neurol. Sci., 1990, 96:19-27.
34) Warren K.G., Catz I.:"Purification of autoantibodies to myelin basic protein by antigen specific affinity chromatography from cerebrospinal fluid IgG of multiple sclerosis patients. Immunoreactivity studies with human myelin basic protein". J. Neurol. Sci., 1991, 33:55-62.
35) Hafler D.A., Fox D.A., Manning M.E. e coll.:"In vivo activited T lymphocytes in the peripheral blood and cerebrospinal fluid of patients with multiple sclerosis". N. Eng. J. Med., 1985, 312:1405-1411.
36) Tournier-Lasserve E., Hashim G.A., Bach M.A.:"Human T-cell response to myelin basic protein in multiple sclerosis patients and healthy subjects". J. Neurol. Sci. Res., 1988, 19:149-156.
37) Canal N., Comi G., Filippi M.:"La sclerosi multipla: clinica, diagnosi e terapia". Atti del Convegno "Approccio pratico alla sclerosi multipla". Ex Arte Salus - Società Medico-Chirurgica Roveretana, 1992, 178:18-31.
38) McAlpine D.:"Symptoms and signs (chapters 5 and 6)" in McAlpine D., Lumsden C.E., Acheson E.D. (eds):"Multiple sclerosis: a reappraisal". Baltimore, Williams and Wilkins, 1972.
39) Confavreux C., Aimard G., Devic M.:"Course and prognosis of multiple sclerosis assessed by the computerised data processing of 349 patients". Brain, 1980, 103:281-300.
40) Kurtzke J.F., Beebe G.W., Nagler B. e coll.:"Studies on the natural history of multiple sclerosis. 8. Early prognostic features of the later course of the ilness". J. Chronic Dis., 1977, 30:819-830.
41) Charcot J.M.:"Lectures on the disease of the nervous system". Vol. 1. London, 1877.
42) Schumacher G.A., Beebe G., Kubler R.F. e coll.:"Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of the experimental trials of therapy in multiple sclerosis". Ann. NY Acad. Sci., 1965, 122:522-568.
43) McDonald I.W., Halliday A.M.:"The diagnosis and classification of multiple sclerosis". Br. Med. Bull., 1977, 33:4-8.
44) Bydder G.M., Steiner R.E., Young I.R. e coll.:"Clinical NMR imaging of the brain: 140 cases". AJR, 1982, 139:215-236.
45) Comi G., Martinelli V., Filippi M. e coll.:"Paraclinical and laboratory tests in suspected multiple sclerosis at the onset of the disease". Neurology, 1990, 40 (suppl. 1):225.
46) Martinelli V., Comi G., Filippi M. e coll.:"Paraclinical tests in acute-onset optic neuritis: basal data and results of a short follow-up". Acta Neurol. Scand., 1991, 84:231-236.
47) Filippi M., Martinelli V., Locatelli T. e coll.:"Acute and insidious-onset myelopathy of undetermined aetiology: contribution of paraclinical tests to the diagnosis of multiple sclerosis". J. Neurol., 1990, 237:171-176.
48) Lee K.H., Hashimoto S.A., Hooge J.P. e coll.:"Magnetic resonance imaging of the head in the diagnosis of multiple sclerosis: a prospective 2-years follow-up with comparison of clinical evaluation, EPs, oligoclonal banding and CT". Neurology, 1991, 41:657-660.
49) Kermode A.G., Thompson A.J., Tofts P. e coll.:"Breakdown of BBB precedes symptoms and other magnetic resonance imaging signs of new lesions in multiple sclerosis: pathogenetic and clinical implications". Brain, 1990, 113:1477-1489.
50) Hawkins C.P., Munro P.M.G., MacKenzie F. e coll.:"Duration and selectivity of blood-brain barrier breakdown in chronic relapsing experimental allergic encephalomyelitis studied by gadolinium-DTPA and proteins markers". Brain, 1990, 113:365-378.
51) Katz D., Taubenberger J., Raine C. e coll.:"Gadolinium-enhancing lesions on magnetic resonance imaging: neuropathological findings". Ann. Neurol., 1990, 28:243.
52) Thompson A.J., Kermode A.G., MacManus D.G. e coll.:"Patterns of disease activity in multiple sclerosis: clinical and magnetic resonance imaging study". Br. Med. J., 1990, 300:631-634.
53) Thompson A.J., Kermode A.G., Wicks D. e coll.:"Major differences in the dynamics of primary and secondary progressive multiple sclerosis". Ann. Neurol., 1991, 29:53-62.
54) Chiappa K.H.:"EPs in clinical medicine". New York, Raven Press, 1983.
55) Comi G., Galardi G., Filippi M. e coll.:"Magnetic stimulation of motor cortex in multiple sclerosis: correlations with clinical, SEP and brain magnetic resonance imaging findings" in Gonsette R.E., Delmotte P. (eds.):"Recent advances in multiple sclerosis therapy". Elsevier Science Publisher, 1989, p. 309.
56) Poser C.M., Paty D.W., Scheinberg L. e coll.:"New diagnostic criteria for multiple sclerosis: guidelines for research protocols". Ann. Neurol., 1983, 13:227-231.
57) Sharief M.K., Thompson E.J.:"The predictive value of intrathecal Ig synthesis and magnetic resonance imaging in acute isolated syndromes for subsequent development of multiple sclerosis". Ann. Neurol., 1991, 29:147-151.
58) Sharief M.K., Hentges R.:"Importance of intrathecal synthesis of IgD in multiple sclerosis: a combined clinical, immunologic and magnetic resonance imaging study". Arch. Neurol., 1991, 48:1076-1079.
59) McDonald W.I.:"Sclerosi multipla: ottimismo in campo diagnostico". Br. Med. J. (ed. ital.), 1992, vol. 17, 81:110-112.
60) Webb H.E.:"Sclerosi multipla: pessimismo in campo terapeutico". Br. Med. J. (ed. ital.), 1992, vol. 17, 81:112-113.
61) Weinstock-Guttman B., Ransohoff R.M., Vinkel R.P., Rudick R.A. :"The interferons: biological effects, mechanisms of action, and use in multiple sclerosis". Ann.Neurol., 1995, 37(1):7-15.
62) Yednock T.A., Cannon C., Fritz L.C. e coll.:"Prevention of experimental autoimmune encephalomyelitis by antibodies against 4ß1 integrin". Nature, 1992, 356:63-66.
63) Mossetti A., Leveghi D., Corona A.M., Bortolotti P.:"Nuove tecniche di riabilitazione nella sclerosi multipla". Atti del Convegno "Approccio pratico alla sclerosi multipla". Ex Arte Salus -Società Medico-Chirurgica Roveretana, 1992, 178:32-35.
64) Mertin J. :"Rehabilitation in multiple sclerosis". Ann.Neurol., 1994, 36 (Suppl):130-133.
Allegato 1. Scala funzionale di Kurtzke, gradi di compromissione dei diversi sistemi funzionali. (continua)
1) Funzioni piramidali
·
0 = Normale.·
1 = Segni anormali senza disabilità.·
2 = Disabilità minima.·
3 = Lieve o moderata para/emiparesi (ipostenia evidenziabile, ma la funzione è preservata per brevi periodi); marcata monoparesi (abolita ogni funzione).·
4 = Marcata para/emiparesi (la funzione motoria è difficoltosa) o moderata tetraparesi (funzione compromessa, ma mantenuta per brevi periodi) o monoplegia.·
5 = Para/emiplegia o marcata tetraparesi.·
6 = Tetraplegia2) Funzioni cerebellari
·
0 = Normale.·
1 = Segni anormali senza disabilità.·
2 = Lieve atassia (tremori o movimenti impacciati facilmente riconoscibili, ma senza interferenza sulla funzione).·
3 = Moderata atassia (tremori o movimenti impacciati facilmente riconoscibili, ma con modesta interferenza sulla funzione).·
4 = Marcata atassia (funziona gravemente compromessa).·
5 = Gravissima atassia (incapacità a svolgere movimenti coordinati).·
9 = Non noto.Indicare con X quando l'ipostenia (grado 3 o maggiore della funzione piramidale) interferisce
sulla funzione cerebellare).
3) Funzioni del tronco encefalo
·
0 = Normale.·
1 = Segni anormali asintomatici ( p. es.: nistagmo di I° grado).·
2 = Lieve disabilità (p. es.: lieve ipostenia dei muscoli oculomotori o di altro nervo cranico, nistagmo di II° grado, lieve disartria).·
3 = Nistagmo severo (III° grado o marcata ipostenia dei muscoli extraoculari o modesta disfunzione degli altri nervi cranici o modesta disartria).·
4 = Marcata disartria o altra marcata disabilità.·
5 = Impossibilità a deglutire o a parlare.·
9 = Non noto.4) Funzioni sensitive
·
0 = Normale.·
1 = Ipopallestesia o altro difetto in uno o due arti.·
2 = a) Lieve ipoestesia tattile o dolorifica, ipobatiestesia (s. di posizione) e/o ipopallestesia (s. vibratoria) in uno o due arti; b) ipopallestesia in tre o quattro arti.·
3 = a) Moderata ipoestesia tattile, dolorifica o ipobatiestesia e/o ipopallestesia in uno o due arti; b) lieve ipoestesia tattile e dolorifica e/o ipoestesia propriocettiva in tre o quattro arti.·
4 = a) Marcata ipoestesia tattile o dolorifica o propriocettiva, sole o in combinazione in uno o due arti; b) moderata ipoestesia tattile e dolorifica e/o grave difetto della propriocettiva in almeno due arti.·
5 = Anestesia in uno o due arti o moderata ipoestesia tattile o dolorifica e/o difetto della propriocettiva al di sotto del capo.·
6 = Anestesia al di sotto del capo.·
9 = Non noto.5) Funzioni sfinteriche
·
0 = Normale.·
1 = Lieve esitazione o ritenzione o urgenza della minzione.·
2 = Moderata difficoltà ad iniziare il mitto o ad evacuare; ritenzione od urgenza nella minzione o evacuazione (compressione anche manuale per urinare o evacuare; cateterizzazione intermittente) rara incontinenza urinaria.·
3 = Frequente incontinenza urinaria.·
4 = Quasi costante cateterizzazione e costante uso di misure per evacuare.·
5 = Perdita della funzione sfinterica vescicale.·
6 = Perdita di entrambe le funzioni sfinteriche.·
9 = Non noto6) Funzioni visive
·
0 = Visus normale·
1 = Scotoma e visus 7/10.·
2 = L'occhio peggiore presenta scotoma ed acuità visiva massima corretta compresa fra 3/10 e 7/10.·
3 = L'occhio peggiore presenta ampio scotoma, modesto deficit campimetrico ed acuità visiva corretta di 2-3/10.·
4 = a) L'occhio peggiore presenta grave difetto campimetrico ed acuità visiva corretta di 1-2/10. b) come grado 3 e massima acuità visiva corretta di 3-4/10 nell'occhio migliore.·
5 = a) L'occhio peggiore presenta acuità visiva massima di 1/10 o meno; b) come grado 4 e massima acuità visiva corretta nell'occhio migliore di 3-4/10.·
6 = Come grado 5 e massima acuità visiva corretta nell'occhio migliore di 3-4/10.·
9 = Non noto7) Funzioni mentali
·
0 = Normale.·
1 = Lievi alterazioni dell'umore.·
2 = Modesto decadimento mentale.·
3 = Moderato decadimento mentale.·
4 = Marcato decadimento mentale (moderata sindrome cerebrale cronica).·
5 = Demenza o sindrome cerebrale cronica.·
9 = Non noto.8) Altre funzioni
A) Spasticità
·
0 = Assente.·
1 = Lieve.·
2 = Moderata.·
3 = Grave.·
9 = Non noto.B) Ogni altra funzione neurologica attribuibile alla sclerosi multipla.
|
Allegato 2. Scala di invalidità per la sclerosi multipla (Disability Status Scale = DSS). |
· 0 = Grado 0 in tutti i sistemi funzionali - Esame neurologico normale.· 1 = Grado 1 in uno o più s.f. - Nessuna invalidità e minimi segni come quello di Babinski o ipopallestesia.· 2 = Grado 2 in uno o due s.f. - Minima invalidità da ipostenia o andatura incerta, disturbi visivi o sensitivi, ecc.· 3 = Grado 3 in uno o due s.f. oppure grado 2 in tre o più s.f. - Moderata invalidità da monoparesi, atassia moderata o combinazioni di disfunzioni minori, capacità deambulatoria interamente conservata.· 4 = Grado 4 in un s.f. oppure grado 3 in tre o più s.f. - invalidità relativamente grave, sebbene interamente in grado di camminare o autosufficiente ed alzato per circa 12 ore al giorno.· 5 = Grado 5 in un s.f. o combinazioni di gradi minori - Grave invalidità che preclude la capacità di lavoro per tutto il giorno, senza speciali provvedimenti. Massima funzione motoria: camminare senz'aiuto per 100-200 metri.· 6 = Grado 3 o peggiore in combinazione in più s.f. - Assistenza (bastoni, grucce o bretelle) per camminare.· 7 = Grado 4 o peggiore in più s.f.; raramente grado 5 solo nel sistema piramidale - Costretto ad una sedia a rotelle, ma capace di guidare le ruote ed entrare ed uscire da solo dalla sedia.· 8 = Grado 4 o peggiore nella maggior parte dei s.f. - Costretto a letto, ma con effettivo uso delle braccia.· 9 = Grado 4 o più alto nella maggior parte dei s.f. - Pazienti totalmente impotenti a letto.· 10= Morte dovuta a sclerosi multipla. |
|
Allegato 3. Scala di invalidità espansa per la sclerosi multipla (Expanded Disability Status Scale = EDSS). |
· 0.0 = Grado 0 in tutti i sistemi funzionali - Normale.· 1.0 = Grado 1 in un s.f. - Non vi è disabilità.· 1.5 = Grado 1 in più s.f. escluse le funzioni mentali - Non vi è disabilità.· 2.0 = Grado 2 in un s.f., altri di grado 0 o 1 - Disabilità minima.· 2.5 = Grado 2 in due s.f., altri di grado 0 o 1 - Disabilità minima.· 3.0 = a) Grado 3 in un s.f., altri di grado 0 o 1 - Disabilità moderata. b) Grado 2 in tre oquattro s.f., altri di grado 0 o 1 - Disabilità lieve. Il paziente è del tutto autonomo.· 3.5 = a) Grado 3 in un s.f. e grado 2 in uno o due s.f.; b) grado 2 in cinque s.f., altri di grado 0 o 1 - il p. è del tutto autonomo, ma ha una disabilità moderata.· 4.0 = Grado 4 in un s.f., altri di grado 0 o 1 o combinazioni di gradi inferiori che superano i limiti dei punteggi precedenti - Il p. è del tutto autonomo senza aiuto, autosufficiente ed alzato per 12 ore al giorno, è in grado di camminare senza aiuto e senza fermarsi per circa 500 metri.· 4.5 = Grado 4 in un s.f., altri di gradi 0 o 1 o combinazioni di gradi inferiori che superano i limiti dei punteggi precedenti - Il p. è del tutto autonomo senza aiuto, in grado di lavorare per tutto il giorno, anche se può avere qualche limitazione per una attività completa e richiedere un minimo di assistenza; è in grado di camminare senza aiuto e senza fermarsi per circa 300 metri.· 5.0 = Grado 5 in un s.f. o combinazioni di gradi inferiori che superano i limiti del punteggio 4.0 - Il p. è in grado di camminare senza aiuto e senza fermarsi per circa 200 metri; la disabilità è sufficientemente marcata da intralciare una attività lavorativa completa senza provvedimenti particolari.· 5.5 = Grado 5 in un s.f. o combinazioni di gradi inferiori che superano i limiti del punteggio· 4.0 - Il p. è in grado di camminare senza aiuto e senza fermarsi per circa 100 metri; la disabilità è sufficientemente marcata da impedire una attività lavorativa completa.· 6.0 = Grado 3 o superiore in più di due s.f., altri in varia combinazione - Il p. necessita di assistenza saltuaria o costante da un lato (bastone, gruccia, cinghia) per camminare per circa 100 metri fermandosi o no.· 6.5 = Grado 3 o superiore in più di due s.f., altri in varia combinazione - Il p. necessita di assistenza costante bilaterale (bastoni, grucce, cinghie) per camminare per circa 20 metri senza fermarsi.· 7.0 = Grado 4 o superiore in più di un s.f., altri in varia combinazione; raramente grado 5 solo nel sistema piramidale - Il p. è incapace di camminare oltre 5 metri anche con aiuto ed è essenzialmente obbligato ad una sedia a rotelle; è in grado di spostarsi da solo sulla sedia a rotelle e di trasferirsi in altra sede (letto poltrona); passa in carrozzella circa 12 ore al giorno.· 7.5 = Grado 4 o superiore in più di un s.f. - Il p. è incapace di fare più di qualche passo, è obbligato alla sedia a rotelle; può aver bisogno di aiuto per trasferirsi dalla sedia ad altra sede; si sposta da solo sulla carrozzella, ma non può muoversi su una carrozzella manuale per un giorno intero. Può aver bisogno di una carrozzella a motore.· 8.0 = Combinazioni di più s.f. al grado 4 - Il p. è essenzialmente obbligato a letto o su una sedia o viene trasportato sulla carrozzella, ma può stare fuori dal letto per una gran parte del giorno; mantiene molte funzioni di autoassistenza; ha generalmente un uso efficace degli arti superiori.· 8.5 = Combinazioni di più s.f. al grado 4 - Il p. è essenzialmente obbligato al letto per una buona parte del giorno; ha un qualche uso efficace di uno o entrambi gli arti superiori; mantiene alcune funzioni di autoassistenza.· 9.0 = Grado 4 o peggiore in vari s.f. - Il p. è obbligato al letto e dipendente, può solo comunicare e mangiare (viene alimentato).· 9.5 = Grado 4 o peggiore in vari s.f. - Il p. è obbligato al letto e totalmente dipendente; incapace di comunicare efficacemente o mangiare/deglutire.· 10.0= Morte dovuta a sclerosi multipla. |